![]()
上回我们说到,如何实现LAMMPS的安装和(利用串行程序)运行一次LAMMPS。
本文的题目是输出与输入,而不是常见顺序(输入与输出),目的是强调将输出命令的学习顺序放在前面,有利于我们调试程序,从而更好地理解LAMMPS中各命令的作用。
输出命令——dump
本文还是先以Example/colloid/in.colloid为例。
1 | dump 1 all atom 1000 dump.colloid |
“#”是注释的意思。
in.colloid默认把输出都注释掉了,所以没有输出文件。
我们只要把“#”删除,就可以执行对应的命令。
以上命令中出现了三种类型的数据:dump数据文件、jpg格式图片以及mpg格式视频。
最简单的是dump数据文件,也是我们最常用的。通过dump文件我们可以利用其它软件,例如OVITO,很方便的进行数据可视化。效果要比LAMMPS内置的图片和视频输出效果更好。对应的命令是:
1 | dump 1 all atom 1000 dump.colloid |
他的意思其实是:
1 | dump [ID] [group ID] [style] [timestep] [filename] [args] |
ID:为一个dump命令起一个唯一的代号,一般就是1、2、3group ID:之前定义的一组对象的组(group)唯一代号,比如所有原子(all),后者氧原子氢原子。除了默认的group ID外,其它group ID需要在使用之前通过group命令定义。style:数据类型,比如原子三维坐标(atom)。timestep:记录的时间步长,每隔一个timestep就会记录一行数据。filename:输出文件名。这里面的“*”指的是每一个timestep记录一个dump文件,“*”处利用当时的时间步进行替代。args:对应于不同style的一些其它参数,在dump命令有更详细的介绍。
输出文件的格式如下所示:
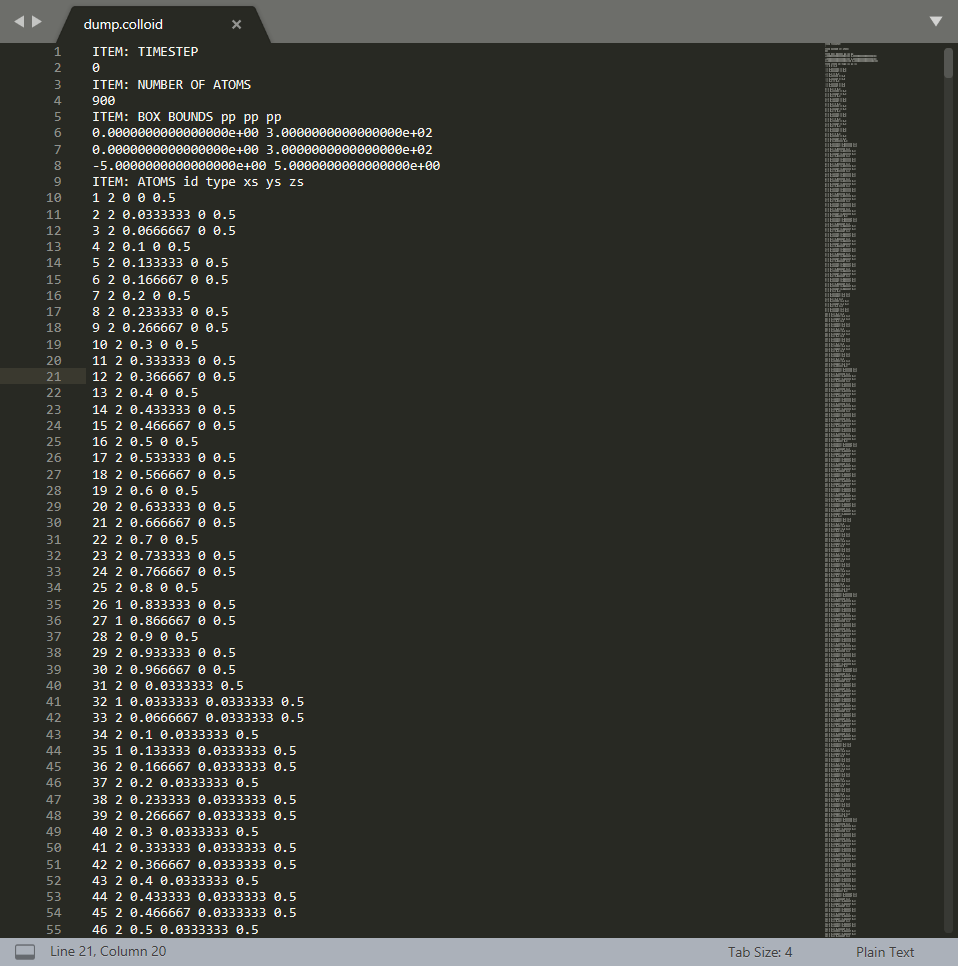
所使用的编辑器为
Sublime Text,相对于记事本,更适合于用来编辑代码和数据,不容易产生未知的符号(编码和文件头问题)。同类竞品有VS Code或Atom,随便下载一个顺手的即可。
可以分析LAMMPS输出的dump文件格式为:

图片或视频的输出类似,不过在args处有其他的参数。博主认为直接输出图片或视频不很必要,感兴趣的读者可以自行了解。另外,dump_modify命令是对之前dump命令中的设置进行的调整,一般是一些默认设置。博主作为新手也不是很了解,感兴趣的读者可以参考官方文档:dump命令和dump_modify命令。
OVITO可视化dump文件是很方便的。在这里博主直接引用一个操作流程。

Ref: http://cms.sjtu.edu.cn/md_manual.html
输入命令read_data
LAMMPS支持lattice命令建立晶体模型,对金属模拟来说是很有用的,在互联网上的教程也很多。博主主要研究的是分子运动的模拟,不属于晶体模拟范畴;而且分子形态和位置都需要自己定义,那么lattice命令就不是那么理想了。接下来博主准备以molecule目录的in.water为例进行LAMMPS数据输入的实例介绍:
1 | LAMMPS根目录\Examples\USER\atc\molecule |
在这个例子中,in.water读取了一个文件water.init。
1 | read_data water.init |
通过LAMMPS运行文件,只要是utf-8编码的文本文件,那么后缀或前缀的不同不会产生太大影响。但一般均按照习惯进行命名。*.init文件也可命名为*.data。我们可以来看看water.init文件的格式。此格式即LAMMPS输入文件的格式。
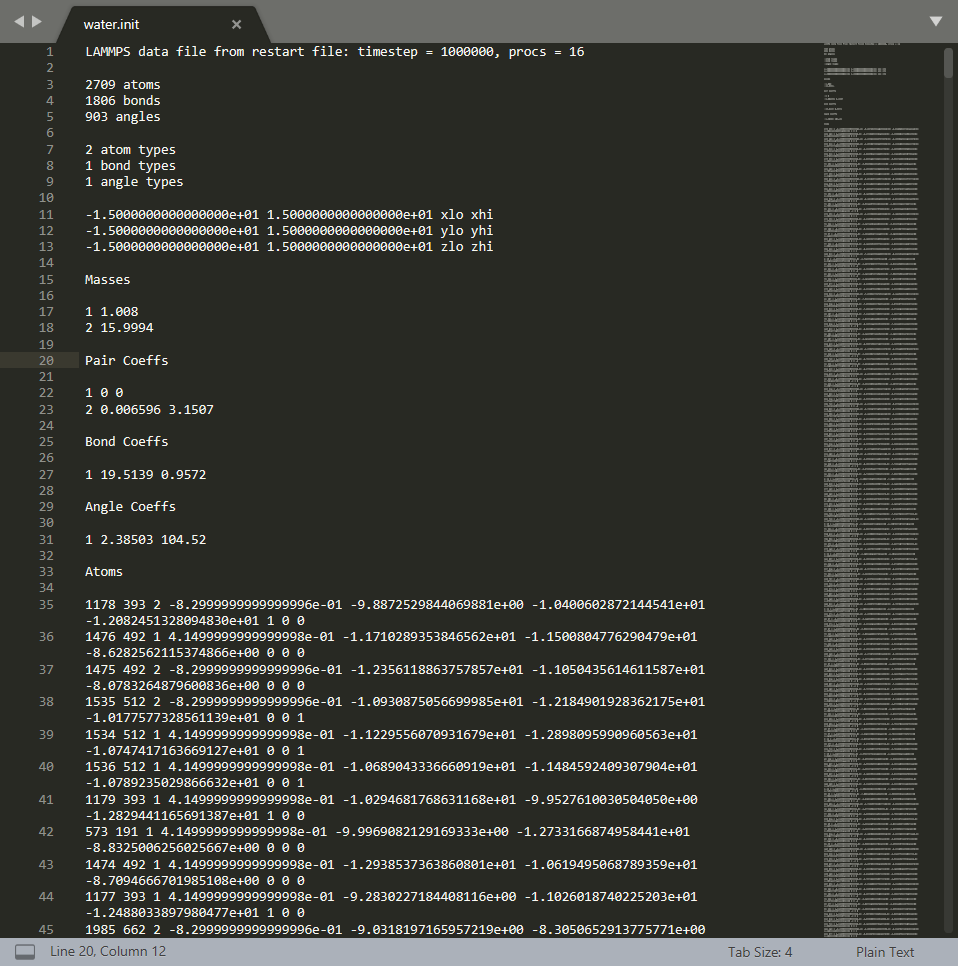
LAMMPS输入文件格式要求较为严格。如果格式不正确将不能正常运行。好在LAMMPS系统做的错误提示比较好用,一般能够根据提示找到输入文件的问题所在。输入文件可以很立体全面地定义输入分子的结构和作用势信息,如原子质量、原子间作用势参数、原子坐标、原子成键等等。读者可以具体参考关于data format的官方链接。
下期预告
到此为止,我们已经是一个可以随意调试脚本并查看调试结果的LAMMPS“脚本小子”了。
如何利用LAMMPS成为真正的盐酒者与磕雪家呢?那其中命令的物理意义是一定要了解的。
博主会挑一些自己用过的命令进行实践和介绍。Love and peace。